Aspergillus niger shows resistance to Zn and Cu; however, limited studies have evaluated the genetic mechanisms underlying metal tolerance in the species. In this study, comparative transcriptome analyses of A. niger F2 under Zn (4000 mg/L) and Cu (3000 mg/L) stress for 15 days were performed to identify genes involved in the response to heavy metal stress. There were more upregulated than downregulated genes under both Cu and Zn stress; however, more genes were differentially expre ssed under Cu than under Zn stress. Downregulated genes under Zn stress were enriched mainly in the membrane part of the cellular component category and for catalytic activity of ribonucleases in the molecular function category. Downregulated genes under Cu stress were enriched for import of Cu ions in the biological process category, intrinsic membrane in the cellula r component category, and reductase and oxidoreductase activity in the molecular function category. Differentially express ed genes under Zn and Cu stress were enriched for different functional domains based on Gene Ontology and Kyoto Ency clopedia of Genes and Genomes analyses. These findings indicated that under heavy metal stress, downregulated genes are mainly involved in ion transport and cell membrane-related functions. Furthermore, energy consumption was higher under Cu stress than under Zn stress, contributing to differences in tolerance levels for A. niger. These findings provide a b asis for genetic engineering for efficient bioremediation.
| Published in | American Journal of Environmental Science and Engineering (Volume 10, Issue 1) |
| DOI | 10.11648/j.ajese.20261001.11 |
| Page(s) | 1-20 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
Aspergillus, Differential Gene Expression, Heavy Metal Stress, Microbial Transcriptome
Item | A1 | A2 | A3 | B1 | B2 | B3 | C1 | C2 | C3 |
|---|---|---|---|---|---|---|---|---|---|
Total Read Count (bp) | 5346231 6 | 5658057 4 | 5825071 4 | 6030647 6 | 5016544 2 | 6225423 4 | 5991535 0 | 37981956 | 4581877 0 |
Total Base | 7799922 | 8232441 | 8539144 | 8842535 | 7338665 | 9109868 | 8757236 | 55362377 | 6765581 |
Count (bp) | 526 | 426 | 295 | 892 | 769 | 248 | 715 | 77 | 236 |
Average Read Length (bp) | 145.9 | 145.5 | 146.59 | 146.63 | 146.29 | 146.33 | 146.16 | 145.76 | 147.66 |
Q10 Base | 7799861 | 8232377 | 8539079 | 8842468 | 7338608 | 9109793 | 8757168 | 55362202 | 6765527 |
Count (bp) | 752 | 865 | 954 | 564 | 593 | 657 | 882 | 13 | 775 |
Q10 Base Ratio (%) | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% | 100.00% |
Q20 Base | 7753259 | 8179973 | 8484155 | 8789274 | 7292127 | 9053973 | 8704413 | 54689303 | 6719240 |
Count (bp) | 917 | 542 | 171 | 716 | 824 | 516 | 955 | 25 | 496 |
Q20 Base Ratio (%) | 99.40% | 99.36% | 99.36% | 99.40% | 99.37% | 99.39% | 99.40% | 98.78% | 99.32% |
Q30 Base | 7594334 | 8003589 | 8297975 | 8607326 | 7135848 | 8862789 | 8522612 | 52872151 | 6562816 |
Count (bp) | 688 | 983 | 402 | 410 | 373 | 155 | 759 | 47 | 477 |
Q30 Base Ratio (%) | 97.36% | 97.22% | 97.18% | 97.34% | 97.24% | 97.29% | 97.32% | 95.50% | 97.00% |
N Base Count (bp) | 60774 | 63561 | 64341 | 67328 | 57176 | 74591 | 67833 | 17564 | 53461 |
N Base Ratio (%) | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
GC Base | 4411946 | 4618611 | 4763806 | 4963075 | 4036200 | 5080221 | 4790932 | 30397402 | 3699814 |
Count (bp) | 663 | 250 | 026 | 619 | 979 | 015 | 607 | 85 | 435 |
GC Base Ratio (%) | 56.56% | 56.10% | 55.79% | 56.13% | 55.00% | 55.77% | 54.71% | 54.91% | 54.69% |
Item | No. | ≥500 bp | ≥1000 bp | N50 | N90 | Max Length (bp) | Min Length (bp) | Total Length (bp) | Average Length (bp) |
|---|---|---|---|---|---|---|---|---|---|
Transcript | 183324 | 108547 | 83291 | 3263 | 719 | 16168 | 201 | 294746548 | 1607.79 |
Unigene | 90345 | 32441 | 20933 | 1876 | 263 | 16168 | 201 | 75544213 | 836.17 |
Item | A1 (%) | A2 (%) | A3 (%) | B1 (%) | B2 (%) | B3 (%) | C1 (%) | C2 (%) | C3 (%) |
|---|---|---|---|---|---|---|---|---|---|
Total reads | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
Total mapped | 97.71 | 97.85 | 97.88 | 97.92 | 97.12 | 97.82 | 97.56 | 98.04 | 97.22 |
Multiple mapped | 81.00 | 81.72 | 82.10 | 79.89 | 81.17 | 79.74 | 83.55 | 83.58 | 81.60 |
Uniquely mapped | 16.71 | 16.13 | 15.78 | 18.04 | 15.95 | 18.07 | 14.01 | 14.46 | 15.62 |
Read-1 mapped | 8.36 | 8.07 | 7.89 | 9.02 | 7.98 | 9.04 | 7.01 | 7.23 | 7.81 |
Read-2 mapped | 8.35 | 8.06 | 7.89 | 9.02 | 7.97 | 9.04 | 7.00 | 7.23 | 7.81 |
Reads map to ‘+’ | 8.35 | 8.07 | 7.92 | 9.02 | 8.00 | 9.03 | 7.03 | 7.25 | 7.85 |
Reads map to ‘- ’ | 8.36 | 8.05 | 7.86 | 9.02 | 7.95 | 9.04 | 6.98 | 7.21 | 7.77 |
Non-splice reads | 16.71 | 16.13 | 15.78 | 18.04 | 15.95 | 18.07 | 14.01 | 14.46 | 15.62 |
Splice reads | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
Reads mapped in proper pairs | 15.41 | 14.85 | 14.71 | 16.79 | 14.74 | 16.71 | 12.96 | 13.55 | 14.51 |
Item | A1 | A2 | A3 | B1 | B2 | B3 | C1 | C2 | C3 |
|---|---|---|---|---|---|---|---|---|---|
0%−10% | 59188 | 57630 | 60157 | 44930 | 50771 | 43478 | 66913 | 71249 | 57765 |
10%−20% | 1750 | 1093 | 1519 | 1897 | 1033 | 1547 | 561 | 2164 | 653 |
20%−30% | 2006 | 1737 | 2045 | 1969 | 1784 | 2185 | 1022 | 1542 | 978 |
30%−40% | 2292 | 2202 | 2198 | 2072 | 2691 | 2863 | 1306 | 1372 | 1395 |
40%−50% | 2662 | 2638 | 2581 | 2357 | 3771 | 3638 | 1831 | 1421 | 2032 |
50%−60% | 3036 | 3051 | 3091 | 2923 | 4323 | 4537 | 2179 | 1506 | 2760 |
60%−70% | 3447 | 3553 | 3454 | 3605 | 5139 | 5113 | 2344 | 1644 | 3347 |
70%−80% | 3494 | 3859 | 3658 | 4290 | 4871 | 5539 | 2522 | 1926 | 3429 |
80%−90% | 4077 | 5132 | 4069 | 6048 | 4868 | 6556 | 3092 | 273 | 3741 |
90%−100% | 8393 | 9450 | 7573 | 20254 | 11094 | 14889 | 8575 | 4788 | 14245 |
 the main resistance mechanism niger under heavy metal stress
the main resistance mechanism niger under heavy metal stress Gene id | Mean TPM (B) | Mean TPM (C) | log2 fold change | q-Value | Direction | Function (KOG/GO/TrEMBL) |
|---|---|---|---|---|---|---|
TRINITY_DN37137 _c4_g3 | 1403.40 39 | 0.0001 | 23.74242701 | 3.54E- 07 | up | Ubiquitin/60S ribosomal protein L40 fusion |
TRINITY_DN35856 _c0_g4 | 951.091 7 | 0.0001 | 23.18115305 | 4.12E- 08 | up | Thioredoxin |
TRINITY_DN37465 _c0_g1 | 808.478 8 | 0.0001 | 22.94677842 | 4.82E- 14 | up | Uncharacterized protein OS=Aspergillus oryzae |
TRINITY_DN37095 _c0_g2 | 550.070 3 | 0.0001 | 22.39118467 | 3.07E- 06 | up | Ubiquitin/40S ribosomal protein S27a fusion |
TRINITY_DN36195 _c1_g9 | 484.202 3 | 0.0001 | 22.20717853 | 9.74E- 08 | up | Copper chaperone |
TRINITY_DN35384 _c0_g2 | 369.312 2 | 0.0001 | 21.81640975 | 1.55E- 05 | up | Molecular chaperones HSP70/HSC70, HSP70 superfamily |
TRINITY_DN35885 _c2_g7 | 307.329 4 | 0.0001 | 21.55135425 | 5.65E- 06 | up | Ubiquitin/60s ribosomal protein L40 fusion |
TRINITY_DN37185 _c0_g3 | 306.433 1 | 0.0001 | 21.54714087 | 1.28E- 07 | up | NADH:flavin oxidoreductase/12- oxophytodienoate reductase |
TRINITY_DN35247 _c0_g4 | 294.128 3 | 0.0001 | 21.48801433 | 0.0005 | up | Glutaredoxin and related proteins |
TRINITY_DN37215 _c0_g1 | 272.012 3 | 0.0001 | 21.37524034 | 2.79E- 07 | up | / |
TRINITY_DN32067 _c0_g1 | 271.879 | 0.0001 | 21.37453329 | 0.0006 | up | 1-Acyl dihydroxyacetone phosphate reductase and related dehydrogenases |
TRINITY_DN36707 _c0_g1 | 235.113 7 | 0.0001 | 21.16492701 | 3.37E- 07 | up | Molecular chaperones HSP70/HSC70, HSP70 superfamily |
TRINITY_DN36887 _c2_g4 | 222.361 | 0.0001 | 21.08447234 | 3.16E- 06 | up | Molecular chaperone (small heat-shock protein Hsp26/Hsp42) |
TRINITY_DN58047 _c0_g1 | 0.0001 | 37.9444 | - 18.53352746 | 0.0111 | down | Subtilisin kexin isozyme-1/site 1 protease, subtilase superfamily |
TRINITY_DN35446 _c3_g1 | 0.0001 | 52.9275 | - 19.01365799 | 0.0115 | down | Integral membrane component |
Gene id | Mean TPM (A) | Mean TPM (C) | log2 fold change | q-Value | Direction | Function |
|---|---|---|---|---|---|---|
TRINITY_DN37048_ c0_g2 | 427.7188 | 0.0001 | 22.0282 | 2.60E- 09 | up | Uncharacterized protein |
TRINITY_DN24487_ c0_g1 | 379.4474 | 0.0001 | 21.8556 | 2.60E- 09 | up | / |
TRINITY_DN35247_ c0_g4 | 313.9668 | 0.0001 | 21.5822 | 2.72E- 06 | up | / |
TRINITY_DN37802_ c1_g1 | 181.5296 | 0.0001 | 20.7918 | 8.50E- 09 | up | Zn2+ transporter ZNT1 and related Cd2+/Zn2+ transporters (cation diffusion facilitator superfamily) |
TRINITY_DN36989_ c0_g9 | 158.1992 | 0.0001 | 20.5933 | 3.26E- 08 | up | oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen |
TRINITY_DN36642_ c0_g6 | 111.8162 | 0.0001 | 20.0927 | 3.08E- 08 | up | oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen |
TRINITY_DN36908_ c0_g3 | 110.6152 | 0.0001 | 20.0771 | 1.65E- 07 | up | / |
TRINITY_DN25819_ c0_g1 | 105.5342 | 0.0001 | 20.0093 | 2.36E- 08 | up | oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen |
TRINITY_DN37305_ c0_g12 | 104.8678 | 0.0001 | 20.0001 | 2.36E- 07 | up | zinc ion binding/oxidoreductase activity |
TRINITY_DN17316_ c0_g2 | 0.0001 | 27.6384 | -18.0763 | 0.0353 | down | cytoplasm/L-amino acid transmembrane transporter activity |
TRINITY_DN37431_ c0_g3 | 0.0001 | 32.7865 | -18.3227 | 0.0169 | down | ribonuclease III activity/RNA binding/RNA processing |
TRINITY_DN22007_ c0_g1 | 0.0001 | 50.1727 | -18.9365 | 9.06E- 05 | down | integral membrane component |
TRINITY_DN35446_ c3_g1 | 0.0001 | 52.9275 | -19.0137 | 0.0002 | down | integral membrane component |
TRINITY_DN25218_ c0_g1 | 0.0001 | 122.6521 | -20.2261 | 0.0002 | down | / |
TRINITY_DN14417_ c0_g1 | 0.0001 | 192.1176 | -20.8736 | 8.44E- 05 | down | Unnamed protein product OS |
TRINITY_DN53309_ c0_g1 | 0.0001 | 861.5829 | -23.03856 | 2.69E- 06 | down | / |
Gene id | Mean TPM (A) | Mean TPM (B) | Mean TPM (C) | log2 fold change | Q-Value | Result 1 | Result 2 |
|---|---|---|---|---|---|---|---|
TRINITY_DN36226_c3_g1 | 911.3373 | 2.4437 | / | 8.5428 | 2.67E-06 | up | up |
TRINITY_DN37865_c1_g4 | 1602.097 9 | 5.7960 | / | 8.1107 | 6.89E-06 | up | up |
TRINITY_DN37487_c0_g1 | 2.6218 | 187.8221 | / | -6.1627 | 0.0093 | down | up |
TRINITY_DN27566_c0_g1 | 1.8571 | 564.1446 | / | -8.2469 | 0.0010 | down | down |
TRINITY_DN30178_c0_g1 | 23.8624 | 0.0001 | / | 17.8644 | 1.0000 | up | up |
TRINITY_DN37711_c1_g5 | 42.2521 | 0.0001 | / | 18.6887 | 2.13E-07 | up | up |
TRINITY_DN37502_c0_g5 | 43.4822 | 0.0001 | / | 18.7301 | 5.95E-05 | up | up |
TRINITY_DN35627_c0_g10 | 37.0141 | 0.0001 | / | 18.4977 | 1.05E-05 | up | up |
TRINITY_DN34499_c2_g1 | 16.9937 | 0.0001 | / | 17.3746 | 1.17E-06 | up | up |
TRINITY_DN35404_c2_g3 | / | 3222.3667 | 10.8374 | 8.2160 | 0.0006 | up | down |
TRINITY_DN37634_c1_g1 | / | 829.3407 | 4.3637 | 7.5703 | 0.0009 | up | up |
TRINITY_DN35878_c1_g1 | / | 6.2588 | 766.4338 | -6.9361 | 0.0109 | down | down |
TRINITY_DN35561_c1_g1 | / | 6.3221 | 647.0381 | -6.6773 | 0.0207 | down | up |
TRINITY_DN35178_c3_g1 | / | 91.5329 | 0.0001 | 19.8040 | 5.84E-06 | up | up |
TRINITY_DN37809_c0_g5 | / | 77.7409 | 0.0001 | 19.5683 | 2.93E-06 | up | up |
TRINITY_DN37260_c1_g3 | / | 53.6735 | 0.0001 | 19.0339 | 1.29E-06 | up | up |
TRINITY_DN36124_c2_g8 | / | 17.6184 | 0.0001 | 17.4267 | 0.0001 | up | up |
TRINITY_DN37249_c1_g12 | / | 29.7571 | 0.0001 | 18.1829 | 7.20E-06 | up | up |
TRINITY_DN37780_c0_g1 | / | 300.2814 | 7.8724 | 5.2534 | 0.0005 | up | up |
TRINITY_DN37780_c0_g1 | 15.8333 | / | 300.2814 | -4.2453 | 0.0046 | down | down |
TRINITY_DN37749_c2_g8 | 5.7350 | / | 60.45897 | -3.3981 | 0.0498 | down | down |
TRINITY_DN33158_c0_g1 | 2.3459 | / | 18.7670 | -2.9999 | 0.0405 | down | up |
TRINITY_DN30925_c0_g1 | 2.0546 | / | 223.2975 | -6.7640 | 0.0004 | down | down |
TRINITY_DN35913_c0_g2 | 315.0660 | / | 3.0729 | 6.6800 | 0.0264 | up | down |
TRINITY_DN37504_c2_g1 | 0.0001 | / | 241.8143 | -21.2055 | 7.26E-07 | down | down |
TRINITY_DN21185_c0_g1 | 0.0001 | / | 95.4216 | -19.8640 | 3.05E-06 | down | down |
TRINITY_DN35275_c1_g2 | 0.0001 | / | 117.9133 | -20.1693 | 7.53E-06 | down | down |
TRINITY_DN36707_c0_g1 | 0.0001 | / | 235.1137 | -21.1650 | 5.16E-07 | down | down |
NCBI | The National Center for Biotechnology Information |
CASAVA Base Calling | Cluster Analysis and Base Calling Application |
CDD | Conserved Domain Database |
TrEMBL | Translation of EMBL Nucleotide Sequence Database |
Swiss-Prot | Swiss Protein Sequence Database |
CDS | Coding Sequence |
RSeQL | Realizability Semantics Query Language |
 This research was supported by the financial support received from the National Natural Science Foundation of China (52304421), Major Project of Changsha Science and Technology Planning Project (kh2301012), Natural Science Foundation of the Environmental Protection Department of Hunan Province (HBKF2022004), and Research Foundation of the Department of Natural Resources of Hunan Province (Grant No. 20230141ST).
This research was supported by the financial support received from the National Natural Science Foundation of China (52304421), Major Project of Changsha Science and Technology Planning Project (kh2301012), Natural Science Foundation of the Environmental Protection Department of Hunan Province (HBKF2022004), and Research Foundation of the Department of Natural Resources of Hunan Province (Grant No. 20230141ST). | [1] | Loa, JDA, Cruz-Rodríguez, IA, Rojas-Avelizapa, NG (2023) Colorimetric detection of metals using CdS-NPs synthesized by an organic extract of Aspergillus niger.Appl. Biochem. Biotechnol. 195, 4148. |
| [2] | Nurdin, M Mulkiyan L, Sugiwati S, Abimayu H, Arifin ZS, Muryanto M, Maulidiyah M, Arham Z, Salim LOA, Irwan I (2023) Productivity of Aspergillus niger InaCC F57 isolate as cellulase agent in OPEFB hydrolysis for glucose high yield. BioNanoScience 13, 114. |
| [3] | Wu L, Zhang L, Li X, Lv R, Cao W, Gao W, Liu J, Xie Z, Liu H(2023) Effective production of kojic acid in engineered Aspergillus niger. Microb. Cell Factories 22, 40. |
| [4] | Amin I, Nazir R, Rather MA (2024) Evaluation of multi-heavy metal tolerance traits of soil-borne fungi for simultaneous removal of hazardous metals. World J. Microbiol. Biotechnol. 40, 175. |
| [5] | El-Mahdy OM, Mohamed HI, Mogazy AM (2021) Biosorption effect of Aspergillus niger and Penicillium chrysosporium for Cd- and Pb-contaminated soil and their physiological effects on Vicia faba L. Environ. Sci. Pollut. Res. Int. 28, 67608. |
| [6] | Xia YT, Guo Q, Qin NN, Tang Y. Tan QJ, Sun HJ, Gao YT, Guo Q, Zhou G(2019) Preparation of Aspergillus niger mycelium-chitosan and study on the adsorption performance of Cr(VI.). Mod. Food Sci. Technol. 35, 68. |
| [7] | Wei BY, Gao GF, Tian Y, Lu XY, Cao L, Li SX,(2011) Decoloring properties of Aspergillus niger to rose bengal and water-soluble color paste. Acta Sci. CIrcumstantiae 31, 492. |
| [8] | Cui H, Liu X, Li K, Cao TT, Cui CW, Wang JY (2019) Mechanism of Hg(II), Cd(II) and Pb(II) ions sorption from aqueous solutions by Aspergillus niger spores. Sep. Sci. Technol. 55, 848. |
| [9] | Deng XH, Chen RH, Zhuo SN, Zhou GY, Shi Y(2019) Bioleaching characteristics of heavy metals from polluted soil with indigenous Aspergillus niger F2. J. Biobased Mater. Bioenergy 13, 401. |
| [10] | Arshadi M, Esmaeili A, Yaghmaei S(2020) Investigating critical parameters for bioremoval of heavy metals from computer printed circuit boards using the fungus Aspergillus niger. Hydrometallurgy 197, 105464. |
| [11] | Bahaloo-HorehN, Mousavi SM, Baniasadi M (2018) Use of adapted metal tolerant Aspergillus niger to enhance bioleaching efficiency of valuable metals from spent lithium-ion mobile phone batteries. J. Cleaner Prod. 197, 1546. |
| [12] | Nguyen TH, Won S, Ha MG, Nguyen DD, Kang HY (2021) Bioleaching for environmental remediation of toxic metals and metalloids: A review on soils, sediments, and mine tailings. Chemosphere 282, 131108. |
| [13] | Hajihoseini J, Fakharpour M (2019) Effect of temperature on bioleaching of iron impurities from kaolin by Aspergillus niger fungal. J. Asian Ceram. Soc. 7, 82. |
| [14] | Kiskira K, Theopisti L, Ioannis L, Lamprini AT, Charalampos P, Konstantina P, Klaus MO, Fotios T, Elias C, · Gerasimos L, Maria OP,(2023) Bioleaching of scandium from bauxite residue using fungus Aspergillus Niger. Waste Biomass Valorization 14, 3377. |
| [15] | Deng XH, Chen RH, Shi Y, Zhuo SN (2018) Preliminary bioleaching of heavy metals from contaminated soil applying Aspergillus niger. Am. J. Environ. Sci. Eng. 2, 72. |
| [16] | Ren WX, Li PJ, Geng Y, Li XJ (2009) Biological leaching of heavy metals from a contaminated soil by Aspergillus niger. J. Hazard. Mater. 167, 164. |
| [17] | Srichandan H, Mohapatrab RK, Parhib PK, Mishraa S (2019) Bioleaching approach for extraction of metal values from secondary solid wastes: A critical review. Hydrometallurgy 189, 105122. |
| [18] | Tripathi S, Poluri KM (2021) Heavy metal detoxification mechanisms by microalgae: Insights from transcriptomics analysis. Environ. Pollut. 285, 117443. |
| [19] | Voigt K, Sharma CM, Mitschke J, Lambrecht SJ, Voß B, Hess WR, Steglich C (2014) Comparative transcriptomics of two environmentally relevant cyanobacteria reveals unexpected transcriptome diversity. ISME J. 8, 2056. |
| [20] | Sun S, Wang Y, He B, Chen J, Leng F, Luo W (2024) Comparative transcriptomics revealed the mechanism of Stenotrophomonas rhizophila JC1 response and biosorption to Pb2+. Environ. Geochem. Health 46, 231. |
| [21] | Wang W, Yan H, Dong G, Li Z, Jiang C, Gu D, Niu D, Zhou D, Luo Y(2022) Comparative transcriptomics reveal different genetic adaptations of biofilm formation in Bacillus subtilis isolate 1JN2 in response to Cd2+ treatment. Front. Microbiol. 13, 1002482. |
| [22] | Kwon MJ, Jørgensen TR, Nitsche BM, Arentshorst M, Park J, Ram AFJ, Meyer V (2012) The transcriptomic fingerprint of glucoamylase over-expression in Aspergillus niger. BMC Genomics 13, 701. |
| [23] | Wang L, Xie Y, Chang J, Wang J, Liu H, Shi M, Zhong Y(2023) A novel sucrose-inducible expression system and its application for production of biomass-degrading enzymes in Aspergillus niger. Biotechnol. Biofuels Bioprod. 16, 23. |
| [24] | Pullan ST, Paul D, Stéphane D, Roger I, Matthew K, Almar N,Jolanda M van M, Raymond W, Martin JB, Sanyasi G, Gregory AT,David BA(2014) RNA-sequencing reveals the complexities of the transcriptional response to lignocellulosic biofuel substrates in Aspergillus niger. Fungal Biol. Biotechnol. 1, 3. |
| [25] | van Munster JM, Jolanda M, van M, Paul D, Stéphane D, Steven TP, Martin JB, Sunir M, Matthew K, Emelie CMN, Kristin W, Richard F, Richard B, Yu XL, Paul D, David BA(2014) The role of carbon starvation in the induction of enzymes that degrade plant- derived carbohydrates in Aspergillus niger. Fungal Genet. Biol. 72, 34. |
| [26] | Novodvorska M, Hayer K, Pullan ST, Wilson R, Blythe MJ, Stam H, Stratford M, Archer DB (2013) Transcriptional landscape of Aspergillus niger at breaking of conidial dormancy revealed by RNA-sequencing. BMC Genomics 14, 246. |
| [27] | Dubey S, Shri M, Gupta A, Rani V, Chakrabarty D (2018) Toxicity and detoxification of heavy metals during plant growth and metabolism. Environ. Chem. Lett. 16, 1169. |
| [28] | Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289. |
| [29] | Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402. |
| [30] | Macomber L, Imlay JA (2009) Theiron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad SciUSA 106, 8344-9. |
| [31] |
Liu J, Tan D, Qiu H, Liang Y, Wu H, Yang Y, Zhao H (2025) Unraveling the stress response and biosorption mechanisms of Aspergillus nigerto rare earth element cerium(III) based on transcriptomics and DNA methylomics. Front. Microbiol. 16, 1674444.
https://d oi.org/10.3389/fmicb.2025.1674444 |
| [32] | Chatterjee S, Das S (2022) Whole-genome sequencing of biofilm-forming and chromium-resistant mangrove fungus Aspergillus niger BSC-1. World J. Microbiol. Biotechnol. 39, 55. |
| [33] | Shi YX, Tang LY, Shao Q, Jiang YZ, Wang ZJ, Peng C, Gu TT, Li Z(2024) The dynamic roles of intracellular vacuoles in heavy metal detoxification by Rhodotorulamucilaginosa. J Appl Microbiol.135, lxae241. |
| [34] | Xu YL, Balaji S, Nanthi B, Binoy S, Yong SO, Zhang W, Cornelia R, Donald S, Mark F, Tony H, Dong ZM (2019) Microbial functional diversity and carbon use feedback in soils as affected by heavy metals. Environ. Int. 125, 478. |
APA Style
Chen, Y., Duan, Y., Xue, T., Deng, X. (2026). Comparative Transcriptomics Reveals the Molecular Mechanism Underlying Heavy Metal Detoxification in Aspergillus Niger. American Journal of Environmental Science and Engineering, 10(1), 1-20. https://doi.org/10.11648/j.ajese.20261001.11
ACS Style
Chen, Y.; Duan, Y.; Xue, T.; Deng, X. Comparative Transcriptomics Reveals the Molecular Mechanism Underlying Heavy Metal Detoxification in Aspergillus Niger. Am. J. Environ. Sci. Eng. 2026, 10(1), 1-20. doi: 10.11648/j.ajese.20261001.11
AMA Style
Chen Y, Duan Y, Xue T, Deng X. Comparative Transcriptomics Reveals the Molecular Mechanism Underlying Heavy Metal Detoxification in Aspergillus Niger. Am J Environ Sci Eng. 2026;10(1):1-20. doi: 10.11648/j.ajese.20261001.11
@article{10.11648/j.ajese.20261001.11,
author = {Yingjie Chen and Yuyuan Duan and Tingfang Xue and Xinhui Deng},
title = {Comparative Transcriptomics Reveals the Molecular Mechanism Underlying Heavy Metal Detoxification in Aspergillus Niger},
journal = {American Journal of Environmental Science and Engineering},
volume = {10},
number = {1},
pages = {1-20},
doi = {10.11648/j.ajese.20261001.11},
url = {https://doi.org/10.11648/j.ajese.20261001.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajese.20261001.11},
abstract = {Aspergillus niger shows resistance to Zn and Cu; however, limited studies have evaluated the genetic mechanisms underlying metal tolerance in the species. In this study, comparative transcriptome analyses of A. niger F2 under Zn (4000 mg/L) and Cu (3000 mg/L) stress for 15 days were performed to identify genes involved in the response to heavy metal stress. There were more upregulated than downregulated genes under both Cu and Zn stress; however, more genes were differentially expre ssed under Cu than under Zn stress. Downregulated genes under Zn stress were enriched mainly in the membrane part of the cellular component category and for catalytic activity of ribonucleases in the molecular function category. Downregulated genes under Cu stress were enriched for import of Cu ions in the biological process category, intrinsic membrane in the cellula r component category, and reductase and oxidoreductase activity in the molecular function category. Differentially express ed genes under Zn and Cu stress were enriched for different functional domains based on Gene Ontology and Kyoto Ency clopedia of Genes and Genomes analyses. These findings indicated that under heavy metal stress, downregulated genes are mainly involved in ion transport and cell membrane-related functions. Furthermore, energy consumption was higher under Cu stress than under Zn stress, contributing to differences in tolerance levels for A. niger. These findings provide a b asis for genetic engineering for efficient bioremediation.},
year = {2026}
}
TY - JOUR T1 - Comparative Transcriptomics Reveals the Molecular Mechanism Underlying Heavy Metal Detoxification in Aspergillus Niger AU - Yingjie Chen AU - Yuyuan Duan AU - Tingfang Xue AU - Xinhui Deng Y1 - 2026/02/11 PY - 2026 N1 - https://doi.org/10.11648/j.ajese.20261001.11 DO - 10.11648/j.ajese.20261001.11 T2 - American Journal of Environmental Science and Engineering JF - American Journal of Environmental Science and Engineering JO - American Journal of Environmental Science and Engineering SP - 1 EP - 20 PB - Science Publishing Group SN - 2578-7993 UR - https://doi.org/10.11648/j.ajese.20261001.11 AB - Aspergillus niger shows resistance to Zn and Cu; however, limited studies have evaluated the genetic mechanisms underlying metal tolerance in the species. In this study, comparative transcriptome analyses of A. niger F2 under Zn (4000 mg/L) and Cu (3000 mg/L) stress for 15 days were performed to identify genes involved in the response to heavy metal stress. There were more upregulated than downregulated genes under both Cu and Zn stress; however, more genes were differentially expre ssed under Cu than under Zn stress. Downregulated genes under Zn stress were enriched mainly in the membrane part of the cellular component category and for catalytic activity of ribonucleases in the molecular function category. Downregulated genes under Cu stress were enriched for import of Cu ions in the biological process category, intrinsic membrane in the cellula r component category, and reductase and oxidoreductase activity in the molecular function category. Differentially express ed genes under Zn and Cu stress were enriched for different functional domains based on Gene Ontology and Kyoto Ency clopedia of Genes and Genomes analyses. These findings indicated that under heavy metal stress, downregulated genes are mainly involved in ion transport and cell membrane-related functions. Furthermore, energy consumption was higher under Cu stress than under Zn stress, contributing to differences in tolerance levels for A. niger. These findings provide a b asis for genetic engineering for efficient bioremediation. VL - 10 IS - 1 ER -
School of Resource & Environment, Hunan University of Technology and Business, Changsha, China
School of Resource & Environment, Hunan University of Technology and Business, Changsha, China
School of Resource & Environment, Hunan University of Technology and Business, Changsha, China
School of Resource & Environment, Hunan University of Technology and Business, Changsha, China
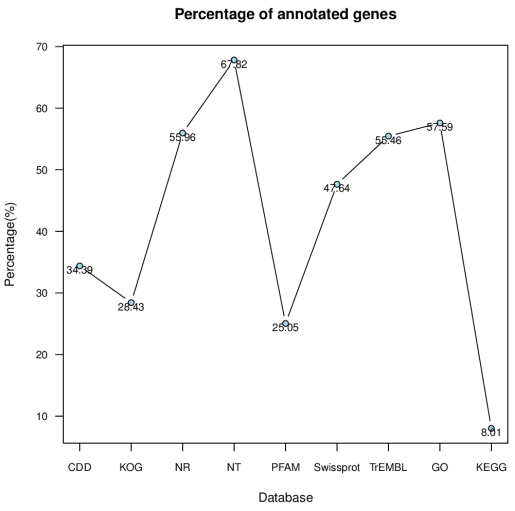
Figure 1. Percentage of annotated genes in different database.
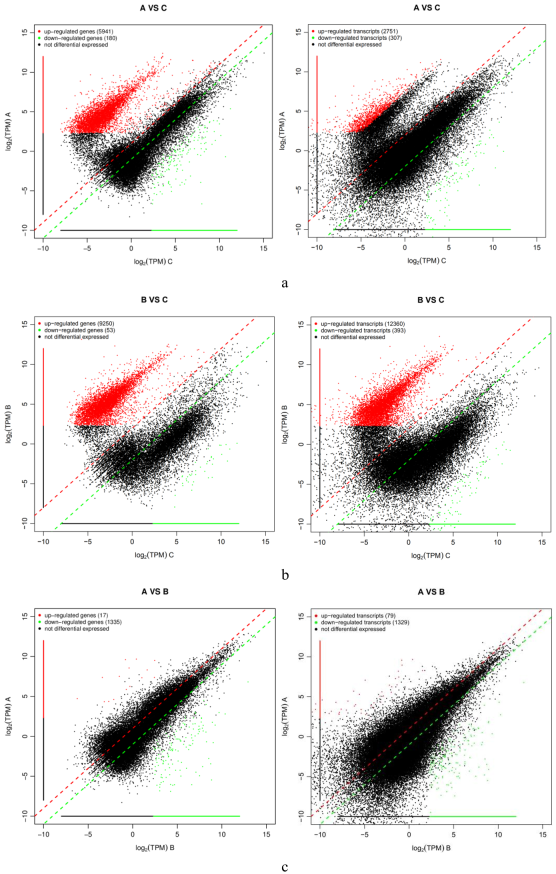
Figure 2. The differential genes under Zn and Cu (qValue<0.05 and |FoldChange|>2).
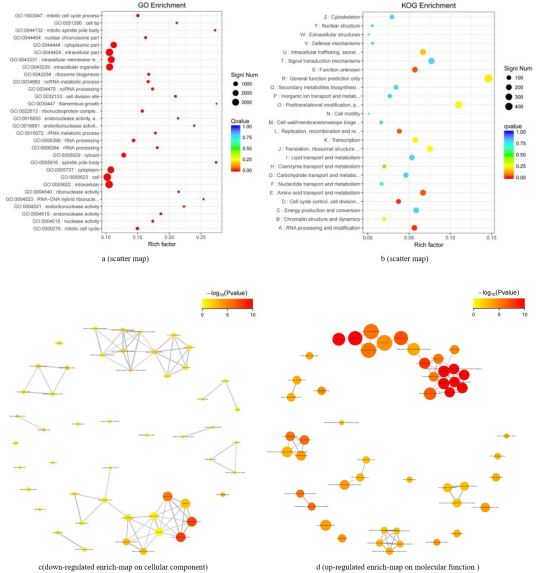
Figure 3. The enrichment of the differential genes (Go database) (A vs C).
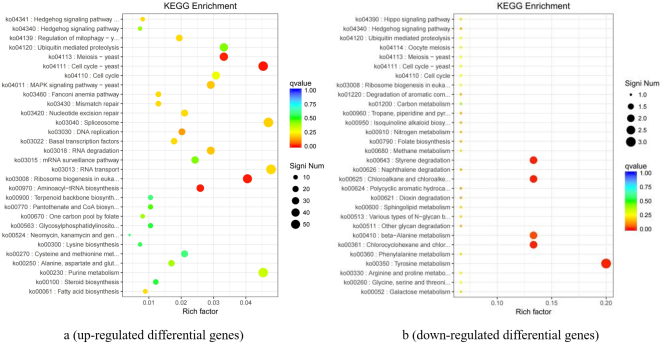
Figure 4. The enrichment of the differential genes (KEGG database)(A vs C).
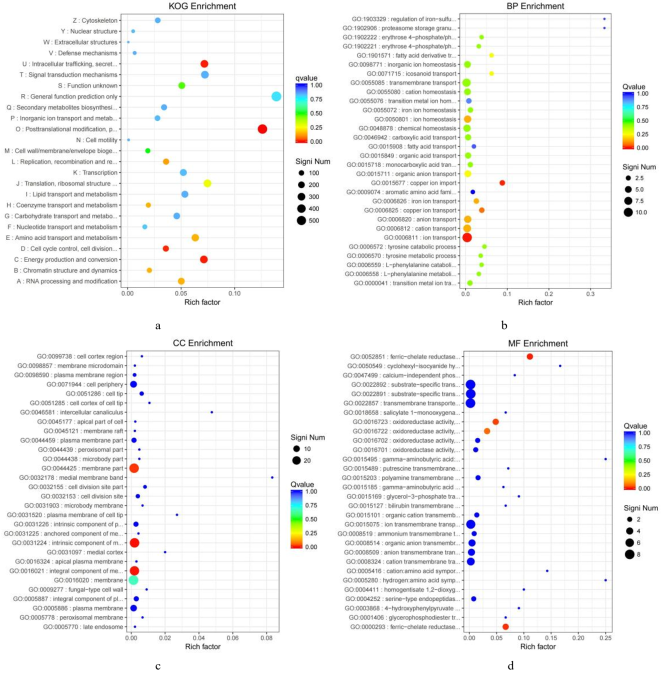
Figure 5. The enrichment of the significantly differential genes (B vsC).
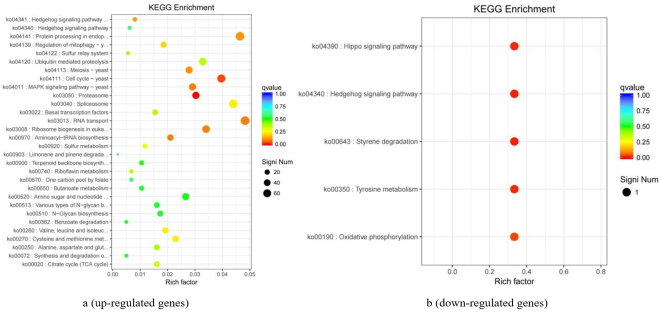
Figure 6. The enrichment of the differential genes (KEGG database) (B vs C).
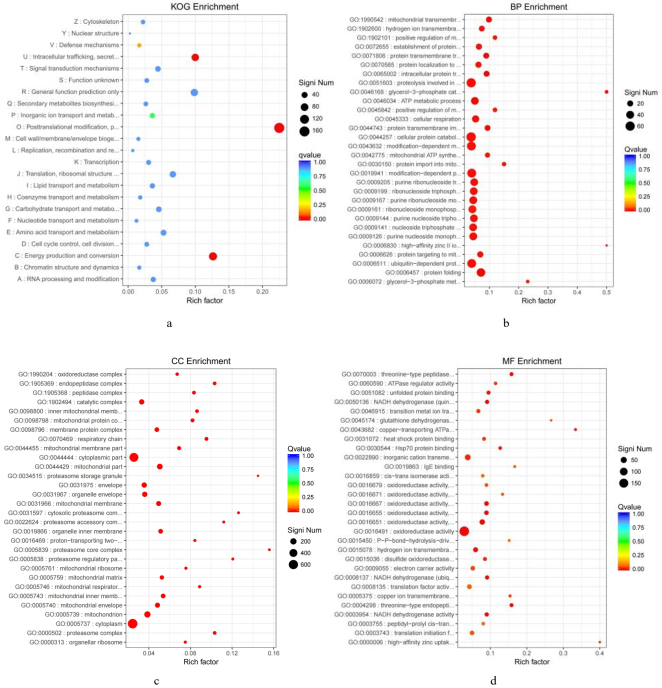
Figure 7. The enrichment of the significantly differential genes (A vs B).
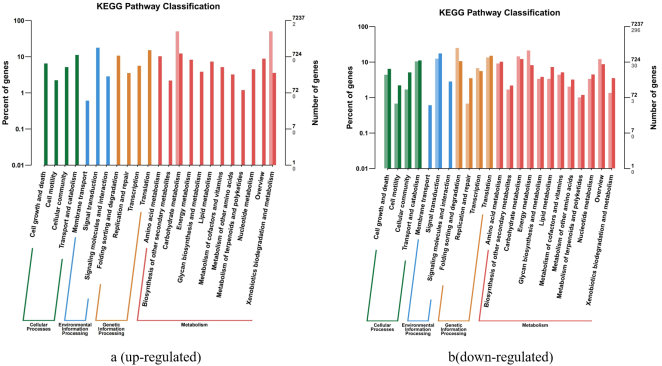
Figure 8. The enrichment of the down-regulated differential genes (KEGG database) (Avs B).
Information

